Course
Chronic Kidney Disease in the Sickle Cell Patient
Course Highlights
- In this Chronic Kidney Disease in the Sickle Cell Patient course, we will learn about the presentation, pathophysiology, and etiology of sickle cell anemia.
- You’ll also learn the presentation of chronic kidney disease in patients with sickle cell anemia.
- You’ll leave this course with a broader understanding of preventative measures and management for chronic kidney disease in patients with sickle cell anemia.
About
Contact Hours Awarded: 1
Course By:
Abbie Schmitt
RN, MSN
Begin Now
Read Course | Complete Survey | Claim Credit
➀ Read and Learn
The following course content
Introduction
Individuals with sickle cell disease are faced with chronic and possibly fatal complications, including chronic kidney disease. This course is designed to explore the pathophysiology of causative factors of sickle cell disease and discuss the impact on kidney function. Nurses will become equipped with knowledge on the clinical signs and symptoms, prevention, and treatment plans for kidney impairment among patients with sickle cell disease.
Sickle Cell Disease
Definition
Sickle cell disease is an umbrella term used for many different types of inherited red blood cell disorders. Those with SCD inherit genetic information that contains a code for abnormal hemoglobin. In sickle cell disease (SCD), red blood cells (RBCs) have a mutation that makes the hemoglobin (Hbg) very sensitive to oxygen changes (11). When a decrease in the oxygen is noticed, the cells begin an observable change from their usual spherical shape to a sickle or crescent shape (11).
Important Terms
- Vaso-occlusive crisis (VOC) or pain crisis: the blockage of blood vessels caused by the abnormal shape of the sickled cells.
- Hemolysis: the breakdown or bursting of red blood cells.
- Anemia: a shortage of red blood cells.
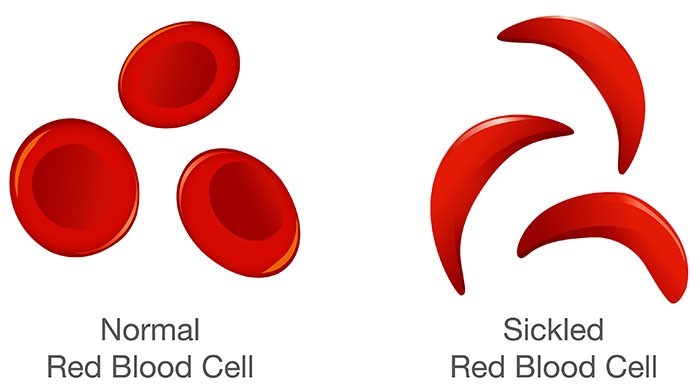
Image 1. Comparison of Normal RBC and Sickled RBC (3)
Epidemiology/Statistical Evidence
Across the world, approximately 3.2 million people are living with SCD, 43 million people have the sickle cell trait (i.e., are carriers of the mutation), and 176,000 deaths are caused by SCD-related complications per year (10). SCD affects approximately 100,000 people in the United States (10).
Statistics show a much higher incidence of SCD among African Americans. Sickle cell anemia is the most common form of SCD in patients of African ethnicity, accounting for approximately 70% of cases (10). The cause of this distribution is unknown. Nearly 10% of African Americans have the sickle cell trait; and one in every 365 African American infants inherit the two sets of SCD genes and develop the disorder (11).

Self Quiz
Ask yourself...
- Can you discuss the difference between a normal red blood cell and a sickled red blood cell?
- How would you explain sickle cell disease to parents of a newborn infant of parents who both have the sickle cell trait?
- Can you list the most common types of SCD?
- Can you name the population that has the highest rate of SCD?
Pathophysiology
Sickle cell disease (SCD) is a form of anemia. Anemia occurs when the body has a low amount of red blood cells (RBCs). RBCs carry hemoglobin (Hgb). Healthy red blood cells are round shaped and easily move through blood vessels to carry vital oxygen to the body. Hemoglobin is an iron-rich protein that carries oxygen from the lungs to all other organs in the body.
In SCD, this abnormal shape of the RBC is now easily broken and tangles in blood vessels and organs, causing clumping and clotting (11). The abnormal shape also results in a decreased lifespan of the RBC. Normal RBCs live around 120 days; sickle cells survival rate is about 10 to 20 days (11).
Symptoms do not usually present in infants until four or five months because the infant is using Hgb made during fetal life, and the sickling process does not affect Hbg at this stage (11).
Disease Types
The most common types of SCD are HbSS, BbSC, and HbS beta thalassemia.
HbSS
- Commonly called sickle cell anemia.
- Usually the most severe form of the disease.
- Two hemoglobin “S” genes inherited, one from each parent.
- Hemoglobin S describes the abnormal form of hemoglobin that causes the red cells to become sickle shaped.
HbSC
- Individual inherits a hemoglobin “S” gene from one parent and a gene for a different type of abnormal hemoglobin called “C” from the other parent.
- This is usually a milder form of SCD.
HbS beta thalassemia
- Individual inherits a hemoglobin “S” gene from one parent and a gene for beta thalassemia, which is another type of hemoglobin abnormality, from the other parent.
Sickle Cell Crisis: happens when a sudden and severe sickling of RBCs causes decreased blood flow in vessels which can lead to tissue necrosis (11). Essentially, this collection of RBCs causes clotting and blockage. This is a life-threatening situation.
Acute Chest Syndrome: in the patient with sickle cell anemia, acute chest syndrome is characterized by fever, leukocytosis, cough, chest pain, and pulmonary infiltrates noted on chest X-rays (6).
Image 2. Normal and sickled red blood cells (7)
Etiology/Causes
SCD is an autosomal recessive hereditary disorder, which means that if both parents pass on the abnormal Hgb, their child will develop the disease (11). However, if only one parent passes it along, the child will have the sickle cell trait and may pass it to their children.
Any condition that causes decreased oxygenation can cause a sickle cell crisis. As we mentioned, the abnormally coded RBC begins to change shape once it senses a change in oxygen saturation. Examples include pneumonia, severe infection, hypothermia, diabetic acidosis, blood loss, or significant dehydration (11).
Many states routinely screen newborns for sickle cell disorder, which is important so that treatment can begin as soon as possible. Early diagnosis and treatment can significantly reduce the risk of complications. Hemoglobin electrophoresis is a blood test that can identify a carrier of sickle cell gene (11).
SCD was first discovered in 1910 (10). Since the discovery, significant strides have been made in understanding pathogenesis of the protean complications, development of targeted molecular therapies, and advancements in treatment plans.
Self Quiz
Ask yourself...
- At what age do SCD symptoms typically present?
- How would you describe the shape of the sickled cells in the X-ray above?
- Would a patient in sickle cell crisis be at higher risk for a blood clot or hemorrhage?
- What triggers the sickle RBC to begin shifting its cell shape?
Sickle Cell Disease and Chronic Kidney Disease
Nurses should become aware of all complications of SCD, including kidney function impairment. Considering the life-sustaining functions of the kidneys, this complication can have disastrous results.
How does SCD affect the kidneys?
Kidney disease is a major clinical concern in sickle cell disorders (SCD). SCD causes kidney problems when sickle cells block blood flow within the kidney and change the way blood vessels work, which reduces oxygen delivery and causes kidney cells to die.
In SCD, anemia can cause an increase in systolic pressure within the kidney, which results in renal hyperperfusion and an increase in glomerular filtration rate (GFR). As a result, renal damage, such as glomerular hypertrophy, can occur. The kidneys are also unable to produce appropriate levels of erythropoietin, which can exacerbate the anemia (9). This cycle can continue until the renal system is severely damaged.
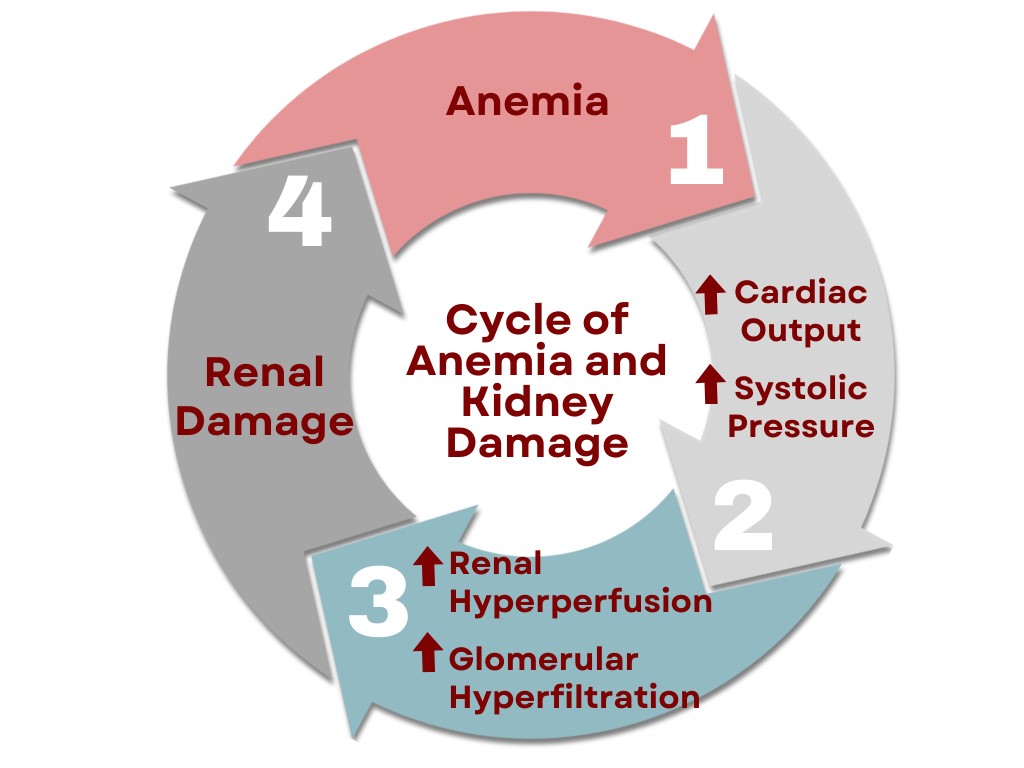
Figure 1. Cycle of Anemia and Kidney Damage. This design was created on Canva.com on December 12, 2023. It is copyrighted by Abbie Schmitt, RN, MSN and may not be reproduced without permission from Nursing CE Central.
Sickle cell nephropathy refers to chronic kidney disease (CKD) that results from the repeated episodes of red blood cell sickling in patients with SCD (6). The clotting and seclusion of vessels reduces blood flow to the kidneys and can lead to chronic kidney disease.
It’s important to consider the function of the kidneys when understanding this complication. Examples of the kidney’s functions include maintaining fluid and chemical homeostasis, production of hormones that help regulate blood pressure and calcium metabolism, and release of a hormone that stimulates red blood cell production (5).
The sickled RBCs harm the glomeruli, leading to glomerulosclerosis, glomerular hypertrophy, interstitial fibrosis, and tissue necrosis (6). The kidney gradually loses the ability to filter out waste, causing proteins that are normally saved by the kidney to be lost in urine (proteinuria).
The oxygen deprivation to the kidneys also leads to the inability to absorb water from urine, leading to hyposthenuria, which is manifested by excessive and nighttime urination. Hematuria is a common finding, resulting from tissue necrosis or other SCD-related complications, such as urinary tract infections and a kidney cancer called renal medullary carcinoma (7). Acute kidney injury (AKI) is common in children and adults with SCD, which causes a sudden rise in blood creatinine levels.
Most people recover kidney function after a single episode, but repeated sickle cell episodes usually lead to chronic kidney disease.
Self Quiz
Ask yourself...
- Can you name some of the major functions of the kidneys?
- How does anemia affect cardiac output and systolic pressure?
- How does increased cardiac output and systolic pressure affect renal perfusion and glomerular filtration?
- Can you describe how impaired kidney function can lead to excessive urination?
Incidence Rates
Sickle cell neuropathy leads to chronic kidney disease in approximately 30% of those with SCD (4). People with sickle cell anemia (HbSS) or sickle beta zero thalassemia have a higher risk than people with other types of SCD. Chronic kidney disease (CKD) is associated with higher risk of mortality. About one in six people with SCD die as a result of chronic kidney disease (4).
Glomerular injury is evident in adults with SCD, with up to 50% of this patient group showing evidence of hyperfiltration (4). The development of end stage renal disease (ESRD) in the adult SCD population has an estimated mean time 37 years from point of SCD development (4).
Most people recover kidney function after a single episode, but repeated sickle cell episodes usually lead to chronic kidney disease.
Clinical Signs and Symptoms
Some of the common findings in patients with sickle cell nephropathy include:
- Renal infarction
-
- Flank or abdominal pain
-
- Nausea and vomiting
-
- Fever
- Hyperkalemia
- Mild hyperchloremic metabolic acidosis
- Urinary tract infections and pyelonephritis
- Nephrogenic diabetes insipidus
- Edema in the legs, ankles, feet, lower abdomen, or face
- Foamy urine
- Weight gain due to fluid retention
- Fatigue and malaise
- Loss of appetite
Some of the laboratory findings in patients with sickle cell nephropathy include:
- Increased Glomerular Filtration Rate (GFR)
-
- The gold standard for assessing how well the kidneys are working
-
- GFR greater than 120 mL/min/1.73 m² is an indicator of abnormal kidney function
-
- Hyperfiltration from glomerular hypertrophy with eGFR of more than 200 ml/minute/1.73 mt2
-
- Hyperfiltration is seen in 51% of patients with HbSS
- Hematuria
-
- Variable presentation: can range from painless microhematuria to visible, painful moderate hematuria.
- Albuminuria
- Proteinuria
- Hyposthenuria
-
- Urine has low specific gravity due to inability of the kidney to concentrate the urine normally
-
- Nearly universal in SCD
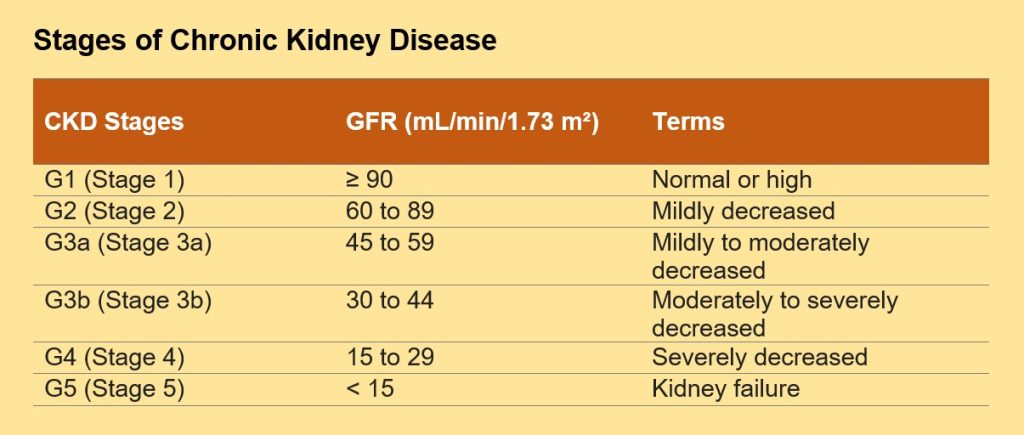
Table 1. Classification of Chronic Kidney Disease Stages (9)
Self Quiz
Ask yourself...
- Can you describe common laboratory findings in a patient with sickle cell disease that has CKD?
- Would you consider the combination of SCD and CKD to have a high mortality rate?
- How would you explain abnormal lab values to a patient with hematuria?
Prevention
Prevention of decreased oxygenation is key in prevention. Predisposing factors for renal injury among SCD patients include exacerbation of anemia, volume depletion, rhabdomyolysis, infections, and the use of non-steroidal analgesics (NSAIDs) (4).
Individuals with SDC should avoid dehydration and avoid extreme exercise. Anemia should be closely monitored and managed. Vaccinations and antibiotics are important to avoid infections. Tobacco cessation is important.
Inhibition of the renin‐angiotensin‐aldosterone system (RAAS) by angiotensin‐converting‐enzyme inhibitors (ACEI) or angiotensin receptor blockers (ARB) can reduce kidney damage by lowering intraglomerular pressure and reducing proteinuria (9).
Studies have suggested that red blood cell transfusions may be beneficial in preventing the progression of kidney disease in children and protective against the onset of microalbuminuria (9).
Treatment
Treatment regimens for CKD in the patient with sickle cell disease incorporates several protocols, including proper hydration, management of infections, RBC transfusion therapy, pain management, treatment of anemia, and bone marrow transplant in some patients. Medications such as Hydroxyurea and ACE-inhibitors are also used for treatment.
Hydration
Hydration helps in the following ways: (4)
- Enhance the osmolality and concentration ability of kidneys.
- Prevent and treat pain crises.
- Intravenous fluids may be required.
- Hypotonic fluid is recommended for renal papillary necrosis.
- Thiazide or loop diuretics are used to maintain urine flow rate (4).
Hydroxyurea
- Classification- Hydroxyurea is an antimetabolite used to treat sickle cell anemia crisis and types of leukemias (2)
- Actions- Hydroxyurea makes your red blood cells bigger. It helps them stay rounder and more flexible to prevent the shape shifting (2).
- Experts strongly recommend hydroxyurea for people with sickle cell disease type HbSS or HbS thalassemia (2).
- Helps reduce the frequency of pain crises and acute chest syndrome.
- The long-term effects of the medication are unknown.
Blood Transfusions
Blood transfusions help by: (9)
- Treating anemia and preventing stroke.
- Diluting the sickled hemoglobin with normal hemoglobin to treat chronic pain, acute chest syndrome, splenic sequestration, and other emergencies.
Vaccinations and Antibiotics
- Prevent and treat infections.
Medications
- Pain medication
- Folic acid (helps prevent severe anemia)
- Urine alkalinization (reduce the acidity of urine in severe cases of hematuria) (1)
- Loop diuretics (to increase urine flow in severe cases of hematuria) (1)
Transplants
- Dialysis or a Kidney Transplant
- Bone marrow transplant (can cure some people with sickle cell disease)
Self Quiz
Ask yourself...
- Can you name the antimetabolite used to treat sickle cell anemia crisis and types of leukemias?
- How does controlling anemia impact the outcomes in SCD?
- Can you discuss ways to prevent significant changes in oxygenation?
Patient Education
Patient education is a vital aspect of advocating for those with SCD and chronic kidney disease. Education on when to seek medical evaluation is important.
The nurse should explain to seek help if they experience fever, shortness of breath, severe head or chest pain, low oxygen saturation, or hypotension. Patients should be aware of their resources and medical team for ongoing care. Consultations will likely include nephrology, hematology, and urology (7).
There is a higher risk for stroke or loss of vision due to the sickling of RBCs. The nurse can provide in-depth education on stroke prevention and recognition of signs and symptoms. Retinal detachment and vision loss can result from blocked or bleeding vessels in the eye from sickled red blood cells (9). The nurse should also coordinate with the healthcare team to make appropriate referrals for evaluation with an ophthalmologist if appropriate.
These individuals more than likely have periods of significant pain. The nurse can help guide their decisions on avoiding drugs that are toxic to the kidneys, such as non‐steroidal anti‐inflammatories (NSAIDS).
Reiteration of the importance of infection prevention measures, hydration, proper diet, and vaccinations is essential to better outcomes. Patients should avoid tobacco, alcohol, and illicit drugs use. Any situation in which oxygen is limited should be avoided, for example, exposure to high altitudes (mountain climbing), exposure to extreme cold or heat, or rigorous exercise (7).
Each treatment plan has specific education topics. Blood transfusion purpose and possible reactions should be discussed with patients. Medication regimen, dosing, purpose, side effects, and complications are also important topics.
SCD is a disease that typically worsens over time, but the nurse can be a source of hope and encouragement to improve quality of life. Resources, encouragement of self-care, support groups, and education are all tools for empowerment. These individuals are living with a life-long condition typically diagnosed at infancy, but it is important to recognize any knowledge deficits and address them in a meaningful method.
Self Quiz
Ask yourself...
- Should you encourage the SCD patient to begin a vigorous exercise regimen?
- Should you encourage the SCD with CKD to take NSAIDS as needed for pain?
- Why would it be important to educate these patients on the signs and symptoms of a stroke?
Conclusion
Nurses can be a meaningful advocate for patients with sickle cell disease who develop chronic kidney disease. Knowledge on the pathophysiology, etiology, and complications of SCD is an important foundation. It is vital to understand how SCD affects the kidneys, recognize clinical manifestations, and be aware of patient-centered prevention and treatment plans for these patients.
References + Disclaimer
- Aeddula NR, Bardhan M, Baradhi KM. Sickle cell nephropathy (2023). In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK526017/
- American Society of Hematology. (2022). Hydroxyurea for sickle cell disease: Treatment information from the American society of hematology. Retrieved from
- Centers for Disease Control and Prevention (CDC). (2023) What is sickle cell disease? Retrieved from https://www.cdc.gov/ncbddd/sicklecell/features/what-is-scd.html
- Erhabor, O. (Ed.). (2022). Sickle Cell Disease. IntechOpen. doi: 10.5772/intechopen.98091
- Hailemariam, F., Falkner, B. (2022). Renal physiology for primary care clinicians. In: McCauley, J., Hamrahian, S.M., Maarouf, O.H. (eds) Approaches to Chronic Kidney Disease. Springer, Cham. https://doi.org/10.1007/978-3-030-83082-3_1
- Mammen, C., Bissonnette, M. L., & Matsell, D. G. (2017). Acute kidney injury in children with sickle cell disease–compounding a chronic problem. Pediatric Nephrology, 32(8), 1287-1291. https://doi.org/10.1007/s00467-017-3650-3
- Mangla A, Ehsan M, Agarwal N, et al. Sickle Cell Anemia (Nursing) (2023). In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK568706/
- Naik RP, Derebail VK. The spectrum of sickle hemoglobin-related nephropathy: from sickle cell disease to sickle trait. Expert Rev Hematol. 2017;10(12):1087-1094. doi:10.1080/17474086.2017.1395279.
- Roy, N. B., Fortin, P. M., Bull, K. R., Doree, C., Trivella, M., Hopewell, S., & Estcourt, L. J. (2017). Interventions for chronic kidney disease in people with sickle cell disease. The Cochrane database of systematic reviews, 7(7), CD012380. https://doi.org/10.1002/14651858.CD012380.pub2
- Sundd, P., Gladwin, M. T., & Novelli, E. M. (2019). Pathophysiology of sickle cell disease. Annual review of pathology, 14, 263–292. https://doi.org/10.1146/annurev-pathmechdis-012418-012838
- Williams, L. S., & Hopper, P. D. (2019). Understanding medical-surgical nursing. F.A. Davis Company.
Disclaimer:
Use of Course Content. The courses provided by NCC are based on industry knowledge and input from professional nurses, experts, practitioners, and other individuals and institutions. The information presented in this course is intended solely for the use of healthcare professionals taking this course, for credit, from NCC. The information is designed to assist healthcare professionals, including nurses, in addressing issues associated with healthcare. The information provided in this course is general in nature and is not designed to address any specific situation. This publication in no way absolves facilities of their responsibility for the appropriate orientation of healthcare professionals. Hospitals or other organizations using this publication as a part of their own orientation processes should review the contents of this publication to ensure accuracy and compliance before using this publication. Knowledge, procedures or insight gained from the Student in the course of taking classes provided by NCC may be used at the Student’s discretion during their course of work or otherwise in a professional capacity. The Student understands and agrees that NCC shall not be held liable for any acts, errors, advice or omissions provided by the Student based on knowledge or advice acquired by NCC. The Student is solely responsible for his/her own actions, even if information and/or education was acquired from a NCC course pertaining to that action or actions. By clicking “complete” you are agreeing to these terms of use.
➁ Complete Survey
Give us your thoughts and feedback
➂ Click the Green MARK COMPLETE Button Below
To receive your certificate