Course
Supporting the Patient in Sickle Cell Crisis
Course Highlights
- In this Supporting the Patient in Sickle Cell Crisis course, we will learn about various clinical manifestations of sickle cell disease.
- You’ll also learn the broad impacts of sickle cell disease on psychological and social well-being.
- You’ll leave this course with a broader understanding of recent advances in the treatment and management of sickle cell disease.
About
Contact Hours Awarded: 3
Course By:
R.E. Hengsterman
MSN, RN
Begin Now
Read Course | Complete Survey | Claim Credit
➀ Read and Learn
The following course content
Introduction
Sickle cell disease (SCD) is a hereditary genetic disorder caused by a variant of the beta-globin gene (β-globin) called sickle hemoglobin (Hb S) that affects hemoglobin (Hb), the part in red blood cells responsible for transporting oxygen throughout the body [1] [11]. In a healthy individual, red blood cells are round and pliable, allowing them to move through the body’s small vasculature. However, in SCD, a genetic defect in the hemoglobin – the protein in red blood cells that carries oxygen – leads to the formation of rigid, sticky (clumped) cells shaped like a sickle, a C-shaped agricultural tool, or the letter C [1]. Their irregular shape obstructs blood flow in small vessels. This blockage can result in episodes of severe pain and heighten the risk of various serious health complications, including infections, acute chest syndrome, and stroke [1].
Sickled cells can accumulate and expire in the spleen, leading to chronic anemia due to a reduction in healthy red blood cell count. The continual presence of sickled cells can cause damage to the spleen itself, increasing the risk of infections [2]. These malformed red blood cells have a shorter lifespan, leading to a persistent deficiency, known as anemia [2]. Average red blood cells have a lifespan of up to 120 days, sickle cells survive for a shorter period, between 10 to 20 days [2].
In 1904, the reported first instance of sickle cell disease (SCD) in the United States involved Walter Clement Noel, a 20-year-old dental student from Grenada [3]. Noel, seeking treatment for anemia and a leg ulcer at Chicago Presbyterian Hospital, met Dr. James B. Herrick (1861-1954), a renowned cardiologist and medicine professor who was first to describe myocardial infarction in 1912 [3]. Dr. Herrick’s intern, Dr. Ernest E. Irons (1877-1959), conducted the first blood tests where he discovered the distinctive sickle-shaped red blood cells in Noel’s blood sample [3].
In November 1910, Dr. Herrick published the first official case study of SCD in medical literature. His study, entitled “Peculiar Elongated and Sickle-Shaped Red Blood Corpuscles in a Case of Severe Anemia,” appeared in the Archives of Internal Medicine (Chicago) [3] [4].
Even though Dr. James B. Herrick reported the first case of SCD in the United States, the condition has a long-standing presence in African and Mediterranean lineage [5]. African medical records from as early as the 1870s referred to the disease using the term “ogbanjes,” meaning “children who come and go,” reflecting the high infant mortality associated with the condition and in Ghana, one family’s history of the inherited disease dates to 1670 [5]. Vernon Mason coined the term “sickle cell anemia” in 1922 [6].
There are two prevailing theories about the origin of the sickle cell disease (SCD) mutation: the multicentric model and the unicentric model coinciding with the adoption of agriculture ~4000–5000 years ago [7]. Recent evidence suggests that the HBB-βS variant, associated with sickle cell disease, originated from a singular source in the central-western region of Africa, near what is now known as Cameroon ~ 7300 years ago, although other sources propose a timeline extending ~ 22,000 years ago [8][9].
A noteworthy aspect of the sickle cell trait is its protective advantage against Plasmodium falciparum malaria [10]. Research shows that the sickle cell trait confers a 60% reduction in overall mortality risk [10]. This protective effect is most pronounced in early childhood, particularly from 2 to 16 months of age in the period that precedes the development of clinical immunity in regions with high malaria transmission rates [10]. The protective effect of the sickle-cell trait in combating severe malaria has led to a widespread occurrence of the sickle-cell mutation (HBB-βS) across various regions in Africa [7].
Patients with Sickle Cell Disease (SCD) often suffer from intense pain, starting from infancy through adulthood [27]. This pain leads to frequent hospitalizations and affects an individual’s quality of life [1][4]. Pain episodes are unpredictable and episodic, ranking among the most severe forms of pain experienced [28]. This pain results from the obstruction of microcirculation by sickled red blood cells (RBCs), leading to ischemia, swelling, tissue death, and potential organ damage [1]. Infants often show pain through irritability, while in older children and adults, the pain can occur anywhere in the body, triggered by factors like infections or environmental changes [1] [2]. Pain episodes can vary in duration and may evolve into chronic pain.

Self Quiz
Ask yourself...
- How do the challenges faced by patients with chronic illnesses including Sickle Cell Disease influence the social interactions and relationships within their families?
- What strategies can families adopt to support or improve their social well-being when a family member is living with a chronic illness?
- In what ways does societal understanding or misunderstanding of chronic illnesses affect the social well-being of patients and their families?
- How crucial is the role of community and social support networks in addressing the social well-being of families affected by chronic illnesses?
Sickle Cell Case Study
Patient Information
- Patient: Jason Keel (pseudonym)
- Age: 9 years
- Sex: Male
- Race: African American
- Past medical history: Notable for asthma, no prior hospitalizations.
- Family history: Both patents with the mutated hemoglobin S gene (HbS)
Presenting Complaint
Patient presents to the emergency department with severe pain in his lower back and legs, which he rates as 9/10 in intensity. The pain started the previous night.
History of Present Illness (HPI)
- The pain is sharp and persistent, not relieved by over-the-counter pain medications (Tylenol).
- No recent injuries, trauma or excessive physical activities could explain the pain.
- The patient also reports feeling fatigued and short of breath.
- No reports of fever, chest pain, or other systemic symptoms.
Physical Examination
- Vitals: BP 120/75 mmHg, Heart Rate 98 bpm, Respiratory Rate 22 breaths/min, Temp 98.6°F, Oxygen Saturation 95% on room air.
- General: Appears in moderate distress due to pain.
- Cardiovascular: Regular rhythm, no murmurs.
- Respiratory: Clear to auscultation bilaterally.
- Abdominal: Soft, non-tender.
- Musculoskeletal: Pain on palpation of the lumbar spine and long bones of the legs. No swelling or redness.
- Skin: Jaundice noted.
Diagnostic Workup
- Complete Blood Count (CBC): Shows anemia with a hemoglobin level of 7 g/dL.
- Peripheral Smear: Presence of sickle cells.
- Hemoglobin Electrophoresis: Confirms HbSS genotype.
- Chest X-ray: Clear, no signs of acute chest syndrome.
- Pain Assessment: Severe pain not controlled by NSAIDs.
Diagnosis
Sickle Cell vaso occlusive crisis.
Treatment Plan
- Pain Management: IV opioids for pain control.
- Hydration: IV fluids to support hydration.
- Oxygen Therapy: Supplemental oxygen to support saturation above 95%.
- Monitoring: Regular monitoring of vital signs and hemoglobin levels.
- Education: Educate the patient and family about sickle cell disease management, including the importance of staying hydrated, avoiding temperature extremes, and seeking prompt medical care for future crises.
Follow-up
- Regular follow-up with a hematologist.
- Discussion about hydroxyurea therapy for chronic management.
- Genetic counseling for the patient and family.
- Vaccinations and prophylactic antibiotics to prevent infections.

Self Quiz
Ask yourself...
- How might the presence of both parents carrying the mutated hemoglobin S gene (HbS) influence the management and long-term treatment strategy for a pediatric patient like Jason Keel?
- Considering Jason’s case of Sickle Cell vaso-occlusive crisis, what role does patient and family education play in managing the disease, preventing future crises, and enhancing the overall quality of life for pediatric patients?
Case Study Discussion
This case highlights a classic presentation of a vaso occlusive crisis (blood vessel occlusion) in a patient with sickle cell disease. Initial patient management focuses on symptomatic management and addressing the immediate crisis [14]. Long-term considerations include managing anemia, preventing infections, and addressing any organ damage due to the disease [14].
To have a child with sickle cell disease (SCD), both parents must carry a faulty gene related to the disorder. SCD is an inherited autosomal recessive pattern. This means that each parent must have one copy of the mutated gene (hemoglobin S gene) and pass it on to their child [12]. When both parents carry one hemoglobin S gene, there is a 25% chance with each pregnancy that the child will inherit two hemoglobin S genes, resulting in sickle cell disease [12]. If a child inherits only one sickle cell gene (hemoglobin S) from one parent and a normal hemoglobin gene (hemoglobin A) from the other parent, the child will have the sickle cell trait [12]. People with the sickle cell trait most often do not have the disease, but they are carriers of the sickle cell gene and can pass it on to their children [13]. The sickle cell trait (SCT) is present in about 1 in every 13 Black or African American newborns [13]. Over 100,000 individuals in the United States live with Sickle Cell Disease (SCD) [13]. The incidence of SCD is about 1 in every 365 births among Black or African American individuals [13]. In the Hispanic American population, SCD occurs at a rate of 1 in every 16,300 births [13].
Children with Sickle Cell Anemia (SCA) often receive a diagnosis during childhood secondary to extensive newborn screening programs in developed nations that can detect the condition in its neonatal stage [58]. By 2007, the United States had implemented nationwide screening for SCA in all states, using methods like high-performance liquid chromatography and isoelectric focusing [29].
Self Quiz
Ask yourself...
- Considering the autosomal recessive inheritance pattern of SCD, how might genetic counseling be beneficial for prospective parents who are known carriers of the sickle cell trait?
- What key information should such counseling provide to help them understand the risks and implications of having a child with SCD?
- Reflecting on the widespread implementation of newborn screening programs for SCA in developed countries, what are the potential benefits and challenges of such programs in terms of early diagnosis and management?
Pathophysiology
The pathophysiology of sickle cell disease (SCD) involves a complex interplay of various processes. Sickle cell disease is a genetic condition affecting hemoglobin production, caused by a specific genetic mutation in the β-globin gene [1]. This mutation results in the formation of an abnormal hemoglobin, known as hemoglobin S (HbS), which is the underlying cause of the disease’s symptoms and complications [15]. This mutation produces abnormal hemoglobin S (HbS), which polymerizes when deoxygenated, causing red blood cells to become misshapen and leading to their premature destruction [16]. This process triggers a chain of events including microvascular vaso-occlusion, ischemia-reperfusion injury, and oxidative stress, resulting in chronic inflammation [16][17]. Increased adhesion of blood cells to vascular endothelium and reduced blood flow contribute to this pathology [18]. In addition, hemolysis, or the breaking down of red blood cells, releases cell-free hemoglobin into the bloodstream, which depletes nitric oxide and worsening endothelial dysfunction [19].
Frequent and unpredictable vaso-occlusive episodes are a defining characteristic of sickle cell disease [20]. A vaso-occlusive crisis arises when sickled red blood cells block the small blood vessels, leading to tissue damage and intense pain due to insufficient blood and oxygen flow to the affected organs [1]. This process involves complex interactions between impaired blood flow, erythrocyte adhesiveness, and blood clotting activation [20]. Factors like blood viscosity, erythrocyte deformability, and exposure of adhesion molecules on erythrocytes, including those from immature reticulocytes, play a significant role [20] [21]. Endothelial dysfunction and inflammation further worsen these issues by increasing cell adhesion and promoting activation of blood cells [21].
In tissues with high oxygen demand, the deoxygenation of intraerythrocytic hemoglobin S (HbS) exposes the hydrophobic groups present on the HbS tetramers (polymer). This results in hydrophobic interactions between the HbS tetramers (polymer) leading to polymerization [22]. These polymers form fibers, increasing cellular rigidity and distorting erythrocyte membranes, resulting in sickling, cellular stress, and hemolysis [22]. The concentration of HbS and fetal hemoglobin (HbF) influences the level of polymerization [22] [23]. Advances in understanding HbS polymerization have led to the development of various therapeutic strategies targeting various stages of sickle cell disease pathobiology [21] [22].
Self Quiz
Ask yourself...
- How does the concentration of HbS and fetal hemoglobin (HbF) in the blood influence the severity and frequency of vaso-occlusive crises and other complications associated with the disease?
- Considering the multifaceted nature of sickle cell disease, involving erythrocyte deformability, blood viscosity, and endothelial dysfunction, how do these factors contribute to the onset of vaso-occlusive episodes?
- Reflecting on the advancements in understanding HbS polymerization and its role in sickle cell disease, what are the most promising therapeutic strategies that have appeared from this knowledge?
- Considering the multifaceted nature of sickle cell disease, involving erythrocyte deformability, blood viscosity, and endothelial dysfunction, how do these factors contribute to the onset of vaso-occlusive episodes?
Etiology
Sickle cell disease (SCD) is a prevalent genetic disorder caused by a specific mutation in the gene responsible for the beta subunit of hemoglobin [4]. Sickle cell anemia (SCA), also known as HbSS disease, is the most severe form of sickle cell disease (SCD). It occurs when an individual inherits two sickle cell alleles (S), one from each parent, making them homozygous for the mutation [4][12]. This genetic configuration results in the specific manifestation of the disease known as HbSS [4].
The misshapen, sickle-shaped red blood cell characteristic of sickle cell anemia (SCA) are inefficient in transporting oxygen and cause blockages in capillaries [4] [12]. This obstruction restricts blood flow to vital organs, including the brain and heart, leading to intense pain due to vaso-occlusion [4]. The majority of symptoms stem from a single nucleotide mutation [25]. SCA stands out as a critical disease caused by a unique change in one of the three billion nucleotides making up the human genome [25].
SCD disease affects multiple organs, caused by a genetic mutation that leads to the replacement of glutamic acid with valine at the sixth position of the beta chain of hemoglobin [4] [25]. Sickle cell disease (SCD) follows an autosomal recessive inheritance pattern. It is a common genetic disorder among African Americans, where its prevalence is about 1 in 500 [24]. About 1 in 12 African Americans carries the mutation for Sickle Cell Disease (SCD), and over 300,000 infants are born with sickle cell anemia (SCA) each year [24].
In sickle-hemoglobin C disease (HbSC), a form of sickle cell disease (SCD), individuals inherit a sickle cell allele (S) from one parent and an abnormal hemoglobin allele (C) from the other [26]. This variant of SCD tends to be less severe compared to others [4][24].
In sickle-β-thalassemia disease, which includes HbS/β+-thalassemia and HbS/β°-thalassemia, an individual inherits a sickle cell allele (S) from one parent and a β-thalassemia allele from the other. The severity of this type of sickle cell disease varies: HbS/β°-thalassemia often results in a severe form, while HbS/β+-thalassemia leads to a milder condition [4] [13].
Sickle cell trait (SCT), also known as HbAS, results from heterozygosity where one parent contributes a normal allele (A) and the other a sickle cell allele (S). Individuals with SCT often do not show symptoms of sickle cell anemia (SCA) and lead normal lives, but they can pass the trait to their children. Screening processes such as newborn screenings, childbirth or blood donations often find individuals with Sickle cell trait (SCT) [4][13].
Patients with the HbSS phenotype of Sickle Cell Anemia often do not experience classic ‘sickle cell crises’ after birth secondary to the fetal hemoglobin (HbF) still present in their blood, aiding in supporting adequate tissue oxygenation [29]. It takes about 6-9 months for HbF to diminish [29].
Sickle Cell Anemia presents with various phenotypes, not all of which are identical, and these can either co-exist or appear as a range within the disease spectrum [29]. A higher hematocrit (Hct) level compared to other forms of the disease characterizes the vaso-occlusive sub phenotype in Sickle Cell Anemia [29]. This increased Hct leads to greater blood viscosity, which in turn contributes to more frequent vaso-occlusive crises and instances of acute chest syndrome [29].
The hemolysis and vascular sub phenotype in Sickle Cell Anemia show a lower hematocrit (Hct) but elevated levels of lactate dehydrogenase (LDH) and serum bilirubin, display more extensive hemolysis and severe anemia [29] [30]. This subtype has a higher risk of complications such as gallstones, pulmonary hypertension, ischemic stroke, priapism, and nephropathy [29] [30]. Severe anemia increases cardiac workload and organ blood flow, heightening organ damage risk. Increased free heme and hemoglobin in the blood vessels contribute to oxidative damage [30].
In individuals with the high Hb F subtype of Sickle Cell Anemia (SCA), having 10 to 15% levels of fetal hemoglobin (HbF) helps to mitigate the symptoms of the disease [29]. This type of distribution of HbF varies across various parts of the body [29]. The pain-sensitive sub phenotype in Sickle Cell Anemia (SCA) and individual differences in neurophysiology makes some individuals more prone to pain than others [29]. This variation in pain sensitivity means that the experience of pain can differ among individuals with SCA [29].
Epidemiological studies show that around 5.2% of individuals with sickle cell disease experience between three to ten severe pain episodes per year [67] [73]. A pain crisis often resolves in five to seven days, but a more intense crisis can lead to pain lasting weeks or even months [66].
If a vaso-occlusive crisis extends beyond seven days, it is crucial to investigate alternative causes including osteomyelitis, avascular necrosis, or compression deformities [68]. In cases where a bone crisis recurs and persists for weeks, an exchange transfusion may be necessary to break the cycle [69]. Transfusion therapy serves as a critical treatment choice for patients, functioning in both primary and secondary prevention of stroke. Simple and exchange transfusions are employed to manage unrelenting and frequent pain crises, as well as pulmonary hypertension [69]. The frequency, severity, location, and duration of pain crises vary even within the same disease subtype [60]. Those with homozygous sickle cell and sickle cell–β°-thalassemia may experience more frequent vaso-occlusive pain crises compared to individuals with hemoglobin SC and sickle cell–β+-thalassemia genotypes [70]. A complex interplay of genetic, rheological, hematological, microvascular, and endothelial factors influences the severity of the disease [70].
Self Quiz
Ask yourself...
- Given that a specific mutation causes sickle cell anemia (SCA) leading to hemoglobin S (HbS), how does this mutation contribute to the distinct pathophysiological features of SCA, such as vaso-occlusive crises and hemolytic anemia?
- Considering the various phenotypes of Sickle Cell Anemia, such as the vaso-occlusive and hemolysis-vascular subtypes, how do these different presentations affect the clinical management and prognosis of patients with SCA?
- Reflecting on the role of fetal hemoglobin (HbF) in mitigating symptoms in individuals with SCA in the high HbF subtype, how does the presence and concentration of HbF influence the clinical severity of SCA?
- Given the varying frequencies, severities, and durations of pain crises among individuals with different genotypes of sickle cell disease, how might this variability influence the approach to pain management and overall treatment in these patients?
Clinical Signs and Symptoms
Individuals with Sickle Cell Anemia (SCA) may experience a range of acute or chronic complications due to the condition. A prevalent acute complication in SCA is the Vaso-occlusive crisis (VOC) [29]. Every individual with Sickle Cell Anemia (SCA) will experience a Vaso-occlusive crisis (VOC) at some point. One of the earliest manifestations in children, often as young as six months, is dactylitis [24]. Vaso-occlusive crisis (VOC) associated with Sickle Cell Anemia (SCA) can affect any organ in the body, including the head and eyes, but often involves the extremities and chest. When the characteristics of VOC pain seem atypical, it is important to collect a detailed history to exclude other potential causes [24] [29].
In Sickle Cell Disease (SCD), symptomatic anemia is a prevalent symptom in Sickle Cell Anemia (Homozygous S) often causing the lowest hemoglobin levels [29]. Asymptomatic patients have varying steady-state hemoglobin levels based on their specific SCD phenotype, with levels ranging from 60–80 g/L in more severe forms to 100–110 g/L in milder variants [25].
Parvovirus B19, known for causing the fifth disease, is a trigger for Acute Aplastic Crisis in Sickle Cell Disease (SCD) [31]. Parvovirus B19 exacerbates anemia through the temporary suppression of bone marrow erythropoietic activity [31]. In healthy children, this infection is often mild, marked by malaise, fever, and light rash. However, in Sickle Cell Disease (SCD), the impact is more severe due to the virus targeting and destroying red blood cell progenitors in the bone marrow, which impedes new red blood cell production [31]. In healthy individuals this infection resolves within a week. In SCD patients, they often experience a substantial decrease in hemoglobin levels and may require blood transfusions. A rapid drop in Hb at least 3 to 6 gm/dL below the baseline defines an aplastic crisis (Compromised RBC production within the bone marrow) [31].
Individuals with Sickle Cell Disease (SCD) have increased vulnerability to infections, (bacterial sepsis and malaria) in children under five [32]. Children under five with Sickle Cell Disease (SCD) face a risk of infection that is thirty to a hundred times greater than that of healthy children [32]. This elevated risk makes them more susceptible to invasive pneumococcal diseases such as pneumonia, meningitis, and septicemia [32]. In individuals with Sickle Cell Disease, respiratory infections may lead to the development of sickle-cell acute chest syndrome, a serious condition with a significant risk of mortality.
Several factors contribute to the increased infection risks in individuals with Sickle Cell Disease, such as functional asplenia or hyposalemia diminishing splenic immune functions, along with impaired complement fixation, weakened neutrophil activity, and less effective antibody responses [34]. Primary concerns include Streptococcus pneumoniae infections, with other serious infections often caused by Haemophilus influenzae, Neisseria meningitides, and Salmonella strains, the latter leading to osteomyelitis [32].
Splenic Sequestration Crisis in Sickle Cell Disease involves the spleen trapping sickled red blood cells, leading to hypoxia and further cell sickling [35]. This can either be hepatic or splenic sequestration. This cycle causes spleen enlargement and potential sudden circulatory failure due to blood pooling [35]. Symptoms include abdominal distension, weakness, thirst, rapid heartbeat, and breathing difficulties [35]. Splenic sequestration, a serious complication of sickle cell anemia affecting young children and characterized by a rapid decrease in hemoglobin levels by 2 g/dL along with an enlarged spleen [35] [36]. Individuals with non-SCA sickle cell variants, such as HbSC and HbS-beta+ thalassemia, do not experience ‘auto-splenectomy’ as often, a common occurrence in SCA patients [29]. These individuals are at risk of developing splenic sequestration later in life, often accompanied by chronic splenomegaly and hypersplenism [29].
Hepatic sequestration, occurring in all SCA phenotypes, involves the rapid enlargement of the liver, often without liver enlargement. A significant hemoglobin drop (over 2gm/dL) and liver capsule stretching defines the sequestration [29]. Liver enzyme levels may not rise.
Acute Chest Syndrome (ACS) is a major complication and the leading cause of death in Sickle Cell Anemia (SCA) [33]. ACS mimics pneumonia and is a major cause of illness, hospitalization, and death in those with sickle cell disease. It occurs when sickled cells obstruct blood vessels in the lungs, posing an emergency and potentially life-threatening situation [33]. ACS is a frequent reason for hospital admissions and can occur at the onset or develop during hospitalization for other reasons. Risk factors include prior ACS episodes, asthma, or recent events like surgeries or infections [33]. ACS presents with sudden cough and breathlessness, sometimes accompanied by fever. Diagnosis involves blood counts, chemistries, cultures, and a chest X-ray, which shows new pulmonary infiltrates, a key ACS indicator [33]. CT scans can help diagnose suspected pulmonary or fat embolism cases [33].
Acute stroke, a severe complication of Sickle Cell Anemia (SCA), has seen a reduced incidence due to transcranial doppler (TCD) screenings and primary prevention programs [37]. About 10% of children with SCA experience overt strokes, and 20-35% have silent cerebral infarcts [38]. TCD is less effective for adults [39]. Symptoms include severe headaches, altered mental status, slurred speech, seizures, and paralysis [37]. Immediate neurological consultation and imaging tests like CT scans, followed by MRI/MRA, are essential for diagnosis and treatment [37].
Acute intrahepatic cholestasis (AIC) manifests with sudden right upper quadrant pain and characterized by worsening jaundice, an enlarging and tender liver, clay-colored stools, fever, and leukocytosis [29]. Laboratory tests often reveal high bilirubin levels, elevated alkaline phosphatase, and coagulopathy [40]. AIC is a critical medical emergency requiring prompt attention.
Priapism, a prolonged and painful erection lasting over four hours, is a common complication in individuals with Sickle Cell Anemia (SCA), affecting about 35% of all male patients [41]. This condition disrupts normal blood flow and causes persistent erection. Episodes of priapism can lead to fibrosis in the penile tissues resulting in permanent erectile dysfunction [41]. This complication underscores the importance of timely medical intervention during priapic events.
Acute ocular complications in patients with Sickle Cell Anemia (SCA), present unique challenges and require careful consideration. Proliferative Sickle Cell Retinopathy (PSR) is a severe complication of Sickle Cell Disease (SCD) that can threaten vision [45]. It develops due to ischemic events in the retina, which trigger angiogenesis, leading to the formation of new blood vessels in the retina [45].
Hyphema refers to the accumulation of blood within the anterior chamber of the eye, often resulting from blunt trauma [42]. The severity of a hyphema, the extent to which the accumulated blood obscures the cornea, receives grading based on the degree of obscuration [42]. A hyphema can manifest in individuals with both SCA and sickle cell trait [42]. The pathophysiology involves the sickling of red blood cells due to the low oxygen pressure and acidotic nature of the aqueous humor. This sickling process leads to a blockage in the trabecular meshwork, causing a rapid increase in intraocular pressure (IOP) [42].
Patients with SCA are vulnerable to complications arising from high IOP, including the development of Central Retinal Artery Occlusion (CRAO) and secondary hemorrhages [43]. Central Retinal Artery Occlusion (CRAO) is an acute ischemic stroke of the eye, leading to painless loss of vision due to infarction in the retina [44]. CRAO can arise without insult or as a secondary complication to increased IOP from hyphema, Moyamoya syndrome, or acute chest syndrome in patients with SCA [44].
Orbital complications are an uncommon manifestation in sickle cell disease [46]. Orbital Infarction is a rare but serious complication resulting from the infarction of the orbital bone, leading to inflammation and compression of surrounding structures [29]. Patients present with symptoms like proptosis, local pain, and edema of the lid or orbit [29]. Orbital Compression Syndrome (OCS), also known as orbital apex syndrome, is a significant concern. Vision loss due to events occurring at the orbital apex highlights ophthalmoplegia (paralysis or weakness of the eye muscles. [45]. This syndrome can involve cranial nerves II, III, IV, VI, and the first division of CN V [45]. Orbital Compression Syndrome varies in severity, with milder forms often responding to conservative treatment, while severe cases, posing a risk to vision, may require surgical intervention to remove hematomas and protect against vision loss [46].

Self Quiz
Ask yourself...
- Considering the common occurrence of Vaso-occlusive crises (VOC) in individuals with SCA, how do the factors leading to VOC contribute to the diverse manifestations of pain and organ damage observed in different patients?
- Given that SCA patients experience varying steady-state hemoglobin levels based on their specific disease phenotype, how do these levels correlate with the severity and frequency of acute complications such as VOC or Aplastic Crisis?
- Reflecting on the acute aplastic crisis triggered by Parvovirus B19 in SCA patients, how does the virus’s action on red blood cell progenitors exacerbate the underlying anemia of SCA? What implications does this have for the management and prevention of such crises in these patients?
- Considering the increased vulnerability to infections in SCA patients how do the underlying mechanisms related to functional asplenia, and impaired immune responses contribute to this increased risk?
Long-Term Complications
Iron (Fe) overload in Sickle Cell Anemia (SCA) patients often arises from frequent blood transfusions and ongoing hemolysis [48]. Each unit of transfused packed red blood cells introduces an added 200 to 250 mg of iron that can accumulate in the heart, lungs, and endocrine glands [49]. Hepatic cirrhosis due to iron overload is a significant cause of mortality in SCA patients. Studies in thalassemia patients have shown a direct correlation between systemic iron levels, survival rates, and cardiac complications [50].
Avascular Necrosis (AVN) of the head of the femur and humerus is a prevalent cause of chronic pain and disability in Sickle Cell Anemia (SCA) patients, with the hip being one of the most affected joints [51]. AVN arises in bone areas with poor collateral circulation, leading to blockage of capillaries by sickled red blood cells, resulting in hypoxia and bone death [29]. Key risk factors include age, frequency of pain episodes, hemoglobin levels, and alpha-gene deletion [29]. About 50 percent of patients with HbSS develop AVN by the age of 33, with those having HbSS-alpha thalassemia and HbSS-Beta-0 thalassemia facing a higher risk of early onset [29].
Leg ulcers are more prevalent in patients with Sickle Cell Anemia (SCA) than in other sickle cell disease genotypes, affecting 2.5-30% of SCA patients over the age of ten [52]. They are more observed in men and older individuals and less frequent in those with higher total hemoglobin, homozygous genotype [Hb SS; sickle cell anemia (SCA), alpha-gene deletion, and elevated HbF levels [53]. Risk factors include trauma, infections, and severe anemia [29]. Ulcers appear on the medial and lateral aspects of the ankles and vary in size and depth, with chronic ulcers leading to osteomyelitis [29].
Pulmonary Artery Hypertension (PAH) affects 6 to 11% of Sickle Cell Anemia (SCA) patients and falls under the World Health Organization (WHO) Group V classification [54]. Chronic hemolysis in SCA causes pulmonary vascular changes, fitting WHO Group 1 criteria in about 10% of cases [29]. PAH in SCA may also arise from left heart dysfunction, chronic lung disease, chronic thromboembolism, or extra thoracic causes [29]. Symptoms include dyspnea on exertion and leg swelling [54]. Diagnosis involves echocardiography to estimate tricuspid regurgitant jet velocity and right heart catheterization. Elevated TRV and serum NT-pro-BNP levels correlate with higher mortality [29] [45].
Chronic Kidney Disease (CKD) is a major indicator of mortality risk in individuals with Sickle Cell Disease (SCD) [57]. Renal complications in Sickle Cell Anemia (SCA) patients including chronic kidney disease (CKD) and renal failure affect around 30% of adults [57]. The average age at which CKD onset occurred in this group was 37 years [57].
Sickle cell nephropathy (SCN) is a group of renal complications that play a significant role in the mortality of individuals with Sickle Cell Disease (SCD) and causing 16-18% of the deaths in this patient group [55] [56]. Complications and involvement encompass a range of physiologic issues including modified blood flow dynamics, organ enlargement, distinct types of glomerular diseases, chronic kidney disease, acute kidney injury, reduced ability to concentrate urine, distal nephron dysfunction, blood in urine, and heightened susceptibility to urinary tract infections and renal medullary carcinoma [55]. SCN signifies an inherent vascular pathology marked by increased blood flow in the cortex, reduced perfusion in the medulla, and an amplified, stress-provoked response causing narrowing of blood vessels [55].
Sickle Cell Disease (SCD) affects the psychological and social well-being of both patients and their families [59]. In pediatric patients with Sickle Cell Disease (SCD), there are broad impacts on quality of life, psychological and social well-being, daily personal and social interactions, emotional well-being, focus, concentration, and motivation [59]. This impact stems from how pain and other symptoms disrupt daily life, coupled with societal attitudes towards those affected [60]. Cultural influences play a crucial role in these challenges due to specific beliefs and practices [25] [60]. The ability of individuals with SCD to manage their condition differs as there is a wide variation in disease severity, overall health, and quality of life among those affected [25] [59].
SCD leads to a variety of acute and chronic health issues, requiring a collaborative approach that includes a range of medical experts. In the United Kingdom, specialized haemoglobinopathy teams orchestrate comprehensive care for SCD [61].
Self Quiz
Ask yourself...
- Given the prevalence of Avascular Necrosis (AVN) in SCA patients in the hip joint, how do the identified risk factors like age, frequency of pain episodes, and genetic variables contribute to the development and progression of AVN? How might understanding these risk factors inform preventative strategies and early interventions?
- Reflecting on the occurrence of leg ulcers in SCA patients, how do factors such as total hemoglobin levels, genotype, and environmental influences like trauma and infections contribute to the development and chronicity of these ulcers?
- Considering the iron overload in SCA patients is due to frequent blood transfusions and ongoing hemolysis, how does this excess iron contribute to the development of complications like hepatic cirrhosis and cardiac issues?
- Considering the impact of Sickle Cell Disease (SCD) on the psychological and social well-being of pediatric patients, how do pain, symptom management, and societal attitudes influence their quality of life and daily functioning?
Treatment and Management
The treatment of sickle cell anemia focuses on preventing pain episodes, alleviating symptoms, and averting long-term complications [62]. Management involves medications and blood transfusions and for certain children and adolescents. In adults with sickle cell disease (SCD), chronic pain and anemia are the most prevalent complications [62]. Episodic pain impacts quality of life, functional capabilities, and the frequency with which patients with sickle cell disease use healthcare services [64].
Due to concerns about narcotic addiction, tolerance, perceived drug-seeking behavior, excessive sedation, respiratory depression, and lack of specific physical examination findings pain from vaso-occlusive crises in sickle cell disease (SCD) is often undertreated [72]. Data indicates the incidence of opioid addiction in SCD patients is as low as 3 percent [66].
The approach to pain management can differ from one patient to another, influenced by family dynamics, individual pain thresholds, and access to healthcare. Patients can manage episodes of mild to moderate pain at home without needing to visit a healthcare facility [65] [73]. Employing self-help psychological strategies, such as guided imagery, has proven to be an effective adjunct in pain management [65].
Patients who adopt these complementary coping strategies experience fewer hospitalizations [65]. The pain management process involves four stages: assessment, treatment, reassessment, and adjustment [25]. Despite advances in pain management, physicians are often reluctant to give patients adequate dosages of narcotic analgesics because of concerns about addiction, tolerance, and side effects [65].
It is important to recognize a pain crisis early, correct the inciting causes, control pain, maintain euvolemia and, when necessary, administer adequate hemoglobin to decrease the hemoglobin S level [66].
For mild pain, patients can often manage at home with oral fluids and nonnarcotic analgesics. Options include acetaminophen, acetaminophen with codeine or oxycodone, and nonsteroidal anti-inflammatory drugs, unless contraindicated due to conditions like peptic ulcer disease, renal disease, or hepatic dysfunction [73]. In cases of moderate to severe pain, titrated doses of narcotic analgesics can be effective pain control [73].
Opioids, including codeine and oxycodone are best for moderate pain due to side effects like sedation, nausea, and vomiting [73]. Severe pain requiring emergency care or hospitalization requires stronger opioids and patient-controlled analgesia (PCA) [65].
Adjuvant nonopioid agents such as antihistamines and antiemetics can help prevent or relieve opioid-related side effects. Providers may consider Tricyclic antidepressants for chronic pain syndromes resulting from recurrent acute pain crises [73][74].
Nonpharmacologic techniques including physical therapy, rest, heat application, transcutaneous electrical nerve stimulation (TENS), self-hypnosis, and diversional techniques can also be effective [66].
Oxygen therapy in vaso-occlusive crises as routine is unsupported and reserved for patients with hypoxemia [87]. Interpreting pulse oximetry readings in SCD patients requires caution due to differences in oxygen affinity between normal hemoglobin and sickle cell hemoglobin [75].
Transfusion therapy is based on a patient’s hemoglobin levels, with simple transfusion therapy guided by a significant decline from baseline levels. Exchange transfusions are for prolonged refractory vaso-occlusive crises, aiming to reduce hemoglobin S levels to below 20 percent [76] [77]. The goals of transfusion therapy in Sickle Cell Disease (SCD) include both enhancing the oxygen-carrying ability and reducing the proportion of sickle hemoglobin (HbS) in comparison to normal hemoglobin A (HbA) [76][77]. This approach aims to prevent or mitigate the complications associated with vaso-occlusion. In acute scenarios, a simple transfusion can increase oxygen transport ability, but with a risk of increased blood viscosity if the hemoglobin level rises above the patient’s usual baseline. For patients with homozygous HbS (HbSS), the ideal target hemoglobin level is set at 10 g/dL [77].
Providers consider fluid replacement to maintain euvolemia, with oral rehydration for milder crises and parenteral rehydration for severe cases [78]. Large fluid volumes can risk pulmonary edema in patients with renal, cardiac, or pulmonary conditions [78].
Prevention
Strategies to Prevent Pain Crises in Sickle Cell Disease Patients:
- Ensure adequate hydration to prevent dehydration during feverish periods and in hot weather [71].
- Avoid activities like mountain climbing or flying in unpressurized cabins (such as noncommercial flights) at altitudes above 10,000 feet.
- Avoid extreme cold, overexertion during exercise, and drugs that might cause acidosis, such as acetazolamide (Diamox) [88].
- Engage in genetic screening and seek vocational counseling about occupations or participation in intense physical activities (such as military training) in hot environments.
- Avoid hypoxemia during periods of surgery with general anesthesia or when procedures involve hypertonic radiographic dyes.
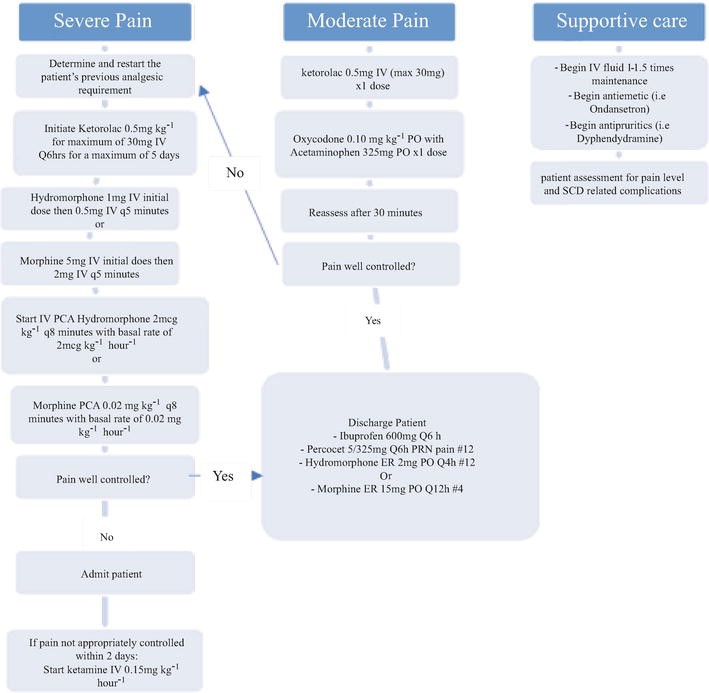
Figure 1. Management of a patient with Sickle Cell Disease (SCD) presenting to the Emergency Department with a Vaso-Occlusive Episode (VOE) [73].
Self Quiz
Ask yourself...
- Given that pain management in SCA is complex and often undertreated due to concerns about narcotic addiction and tolerance, how can healthcare providers balance the need for effective pain relief with the potential risks associated with opioid use?
- Considering the variability in pain management approaches among SCA patients, influenced by factors such as individual pain thresholds and access to healthcare, what are the key considerations in developing a personalized pain management plan for each patient?
- How do nonpharmacologic techniques like physical therapy, heat application, and TENS contribute to pain management in SCA, and in what scenarios might these techniques be beneficial?
- In the context of transfusion therapy for SCA, how do we compare the reduction of the proportion of sickle hemoglobin (HbS) to the goal of enhancing oxygen-carrying ability?
Disease Modifying and Curative Treatments
A stem cell transplant may offer curative treatment in select individuals [62]. Recent and ongoing developments in CRISPR/Cas9 genome editing technology are showing promise as potential cures for individuals with sickle cell disease [62]. In December of 2023, the U.S. Food and Drug Administration (FDA) approved the first gene therapies for treating Sickle Cell Disease (SCD). The two pioneering treatments, Casgevy and Ligeia, for patients aged 12 and older, mark a significant advancement in SCD therapy [63].
For Sickle Cell Disease (SCD), the disease-modifying drugs include hydroxycarbamide and L-glutamine administered daily to decrease the frequency of acute complications, though their effectiveness can differ among individuals [79].
Hydroxycarbamide, also known as hydroxyurea, is a pivotal medication and primary agent in the management of Sickle Cell Disease (SCD) [80]. Hydroxycarbamide’s effectiveness is in its ability to increase overall hemoglobin levels and the production of fetal hemoglobin (HbF) [80]. Hydroxycarbamide also lowers the white blood cell count and reduces the expression of surface adhesion molecules on neutrophils, red blood cells, and vascular endothelium [81]. These actions improve blood flow and lessen vaso-occlusive events.
In July 2017, the U.S. Food and Drug Administration (FDA) sanctioned the use of pharmaceutical-grade L-glutamine for patients with sickle cell disease aged five and above [81]. Clinical trials have shown that this refined form of glutamine lowers the occurrence of acute complications in sickle cell disease [81]. The associated side effects are mild and do not require laboratory monitoring.
Self Quiz
Ask yourself...
- Considering the recent FDA approval of gene therapies such as Casgevy and Ligeia for treating Sickle Cell Disease, how might these new treatments compare to traditional therapies like stem cell transplants in terms of efficacy, safety, and accessibility?
- Given the effectiveness of hydroxycarbamide in increasing fetal hemoglobin (HbF) levels and reducing vaso-occlusive events in SCD, how does its mechanism of action contribute to the overall management of the disease?
- How might the introduction of pharmaceutical-grade L-glutamine, which also lowers the occurrence of acute complications, complement, or change existing treatment strategies for SCD?
Treatments Focused on Decreasing HbS Polymerization
GBT440 (Voxelotor) is an orally administered compound developed to enhance the oxygen affinity of HbS, causing a leftward shift in the oxygen dissociation curve of oxy-HbS [82]. It achieves this by reversibly attaching to the N-terminal valine of the alpha (α) chain of Hemoglobin, altering its structural shape, and stabilizing its oxygenated form [82]. This action leads to a reduction in deoxygenated HbS, which is the variant that polymerizes and results in the sickle cell shape [82].
Nutritional Supplements
Omega-3 fatty acids, extracted and purified from fish oil, provide antioxidant, antithrombotic, and anti-inflammatory effects. The beneficial properties of omega-3 fatty acids include anti-thrombotic eicosanoids, along with a new category of lipid mediators derived from EPA and DHA [83]. In patients with Sickle Cell Disease (SCD), the composition of fatty acids in blood cell membranes is atypical, marked by a high proportion of pro-inflammatory arachidonic acid (AA) compared to the anti-inflammatory fatty acids DHA and EPA, leading to an elevated omega-6/omega-3 ratio [83].
The use of folic acid in Sickle Cell Disease (SCD) may increase red blood cell production and raises the risk of folate deficiency [84]. Folate, also known as vitamin B9, plays a crucial role in the body’s production of red blood cells. Supplementing with folic acid, the synthetic form of folate found in fortified foods and supplements, can help manage the effects of SCD [84].
Self Quiz
Ask yourself...
- Considering the mechanism of action of GBT440 (Voxelotor), which involves enhancing the oxygen affinity of HbS and thereby reducing deoxygenated HbS polymerization, how might this alteration in the oxygen dissociation curve impact the overall pathophysiology of Sickle Cell Disease (SCD)?
- Considering the atypical composition of fatty acids in the blood cell membranes of patients with SCD, characterized by a higher ratio of pro-inflammatory arachidonic acid to anti-inflammatory fatty acids such as DHA and EPA, how might supplementing with omega-3 fatty acids impact the pathophysiology and clinical manifestations of SCD?
- Given the role of folate in red blood cell production and the risk of folate deficiency due to increased red blood cell turnover in SCD, how does supplementing with folic acid support the management of SCD?
Conclusion
Sickle Cell Disease (SCD) is a complex hereditary condition characterized by the presence of sickle hemoglobin (Hb S) that leads to the formation of rigid, sickle-shaped red blood cells [11]. These malformed cells impede blood flow and oxygen transport, causing severe pain, heightened risk of infections, acute chest syndrome, stroke, and chronic anemia [1][11]. First reports of the disease occurred in the United States in 1904 and has a noted prevalence in African and Mediterranean populations [3]. Its protection against malaria has led to a higher frequency of sickle-cell mutation in these regions [7][10] [85].
The management of SCD requires a comprehensive approach, involving hydration, pain control, and the use of various therapeutic agents [78]. The disease’s impact extends beyond physical symptoms, affecting the psychological and social well-being of patients and their families [59]. Cultural factors, individual pain tolerance, and access to healthcare play significant roles in disease management [25][60].
The impact of SCD extends beyond physical symptoms, affecting the psychological and social well-being of patients and their families [59]. Challenges faced by individuals with SCD, and their caregivers include navigating societal misconceptions, cultural beliefs, and healthcare access disparities [86].
Recent advances in SCD research and treatment have opened new avenues for better management of the disease. The development of targeted therapies like GBT440 (Voxelotor), which alters the oxygen affinity of HbS, and nutritional interventions like omega-3 fatty acid supplementation and folic acid, address specific aspects of the disease’s pathophysiology [82] [83] [84]. These advancements, combined with increased awareness and improved healthcare practices, contribute to enhancing the quality of life for individuals with SCD.
Sickle Cell Disease remains a complex and challenging condition, requiring continued research, patient-centric care approaches, and an understanding of the diverse impacts on those affected. The ongoing developments in genetic treatments and comprehensive care strategies hold promise for improving outcomes and providing a more hopeful future for those living with SCD.
Self Quiz
Ask yourself...
Final Reflection Questions
- Considering the significant prevalence of Sickle Cell Disease in African and Mediterranean populations and its evolutionary protection against malaria, how does this geographical and genetic background influence public health strategies and medical interventions for managing SCD in these regions?
- Reflecting on the comprehensive management approach required for SCD, which includes hydration, pain control, and the use of various therapeutic agents, how do cultural factors, individual pain tolerance, and access to healthcare influence the effectiveness of these strategies and the overall quality of life for patients with SCD and their families?
- In the context of splenic sequestration crisis in SCA, how does the trapping of sickled red blood cells in the spleen lead to the acute symptoms observed? How can understanding this mechanism inform the early detection and management of this complication?
- Given the complexity of Acute Chest Syndrome (ACS) in SCA, what are the key pathophysiological processes that differentiate ACS from similar conditions like pneumonia? How does this distinction influence the clinical approach to diagnosis and treatment?
- With the reduced incidence of acute stroke in children with SCA due to transcranial doppler (TCD) screenings and primary prevention programs, how do these strategies address the risk factors for stroke in this population?
- Considering the risk of proliferative Sickle Cell Retinopathy (PSR) and other ocular complications in patients with SCA, how do the unique pathophysiological changes in the eye related to sickle cell disease lead to these complications?
- How should fluid a provider managed fluid replacement in SCA patients to balance the need for maintaining euvolemia with the risk of complications like pulmonary edema, especially in patients with underlying renal, cardiac, or pulmonary conditions?
- Considering the role of oxygen therapy in vaso-occlusive crises, how should healthcare providers decide when to administer oxygen, and what do the potential risks of routine oxygen use in these patients?
- With the emphasis on preventing pain crises in SCA, what lifestyle modifications and preventive measures can patients adopt to minimize the risk of triggering a crisis? How effective are these strategies in real-world settings?
- How do nonopioid adjuvant agents like antihistamines, antiemetics, and tricyclic antidepressants play a role in the overall management of pain in SCA?
- What are the potential benefits and limitations of using omega-3 fatty acids as part of the treatment regimen?
- How does the genetic mutation causing SCD affect the structural integrity of red blood cells and their ability to transport oxygen?
- In what ways does the increased vulnerability to infections in SCD patients impact their overall health and the approach to preventive healthcare?
- Considering the protective effect of the sickle cell trait against malaria, how has this influenced the geographical distribution and prevalence of the sickle cell gene?
- How does the chronic anemia associated with SCD affect the body’s physiological responses and the overall health of patients?
- Given the varying severity of SCD symptoms among individuals, what factors contribute to these differences, and how do they guide personalized treatment plans?
- How does the early childhood onset of SCD symptoms, such as dactylitis, shape the approach to pediatric care and family counseling?
- What are the challenges in managing chronic pain in SCD patients, and how can healthcare providers address concerns about opioid use while ensuring effective pain relief?
- How does the occurrence of acute chest syndrome in SCD patients influence the long-term monitoring and management of their respiratory health?
- What are the implications of chronic organ damage in SCD, such as splenic and hepatic sequestration, for patient prognosis and treatment?
- In the context of SCD, how does the presence of fetal hemoglobin (HbF) affect the clinical outcomes and treatment strategies for patients?
- Considering the psychological and social challenges faced by SCD patients, what support mechanisms are essential for their mental and emotional well-being?
- How do healthcare disparities and cultural factors influence the management and patient outcomes in SCD?
- What role do advancements in genetic research and targeted therapies play in the future landscape of SCD treatment?
- How do complications like priapism and proliferative sickle cell retinopathy in SCD patients require specialized care approaches?
- What challenges remain in reducing the incidence of stroke in adult SCA patients?
- Given the advances in gene editing technologies, what ethical considerations should providers consider in the context of curative treatments for SCD?
References + Disclaimer
- Cleveland Clinic. (2024). Sickle Cell Disease. Retrieved from https://my.clevelandclinic.org/health/diseases/12100-sickle-cell-disease
- Johns Hopkins Medicine. (2024). Sickle Cell Disease. Retrieved from https://www.hopkinsmedicine.org/health/conditions-and-diseases/sickle-cell-disease
- Sebastiani, P., Solovieff, N., Hartley, S. W., Milton, J. N., Riva, A., Dworkis, D. A., … & Baldwin, C. T. (2010). Genetic modifiers of the severity of sickle cell anemia identified through a genome-wide association study. American Journal of Hematology, 85(1), 29-35. Retrieved from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2947974/
- Rare Disease Advisor. (2024). History of Sickle Cell Disease. Retrieved from https://www.rarediseaseadvisor.com/hcp-resource/history-of-sickle-cell-disease/
- Sickle Cell Association. (2021). About Sickle Cell Association St Louis. Retrieved November 4, 2021, from https://sicklecellassociation.org/about-sickle-cell/
- Mason VR. Sickle cell anemia. Journal of the American Medical Association. 1922; 79:1318-1320.
- Esoh K, Wonkam A. Evolutionary history of sickle-cell mutation: implications for global genetic medicine. Hum Mol Genet. 2021 Apr 26;30(R1): R119-R128. doi: 10.1093/hmg/ddab004. PMID: 33461216; PMCID: PMC8117455.
- Shriner, D. and Rotimi, C.N. (2018) Whole-genome-sequence-based haplotypes reveal single origin of the sickle allele during the Holocene wet phase. Am. J. Hum. Genet., 102, 547–556
- Laval, G., Peyrégne, S., Zidane, N., Harmant, C., Renaud, F., Patin, E., Prugnolle, F., Quintana-Murci, L., Laval, G. and Patin, E. (2019) Recent adaptive acquisition by African rainforest hunter-gatherers of the late Pleistocene sickle-cell mutation suggests past differences in malaria exposure. Am. J. Hum. Genet., 104, 553–561
- Centers for Disease Control and Prevention. (2024). Malaria – Biology. Retrieved from https://www.cdc.gov/malaria/about/biology/index.html
- Ashley-Koch A, Yang Q, Olney RS. Sickle hemoglobin (HbS) allele and sickle cell disease: a HuGE review. Am J Epidemiol. 2000 May 1;151(9):839-45. doi: 10.1093/oxfordjournals.aje.a010288. PMID: 10791557.
- National Heart, Lung, and Blood Institute. (2024). Sickle Cell Disease – Causes. Retrieved from https://www.nhlbi.nih.gov/health/sickle-cell-disease/causes
- Centers for Disease Control and Prevention. (2024). What is Sickle Cell Trait? Retrieved from https://www.cdc.gov/ncbddd/sicklecell/traits.html
- Ward R, Simpson E, Verhovsek M. A 19-year-old woman with sickle cell disease and pain. CMAJ. 2016 Jul 12;188(10):745-746. doi: 10.1503/cmaj.150512. Epub 2016 Feb 1. PMID: 26833738; PMCID: PMC4938685.
- Nicola Conran, Carla F. Franco-Penteado & Fernando F. Costa (2009) Newer Aspects of the Pathophysiology of Sickle Cell Disease Vaso-Occlusion, Hemoglobin, 33:1, 1-16, DOI: 10.1080/03630260802625709
- Nader, E., Romana, M., & Connes, P. (2020). The Red Blood Cell-Inflammation Vicious Circle in Sickle Cell Disease. Frontiers in immunology, 11, 454. https://doi.org/10.3389/fimmu.2020.00454
- Conran, N., & Belcher, J. D. (2018). Inflammation in sickle cell disease. Clinical hemorheology and microcirculation, 68(2-3), 263–299. https://doi.org/10.3233/CH-189012
- Rajendran, P., Rengarajan, T., Thangavel, J., Nishigaki, Y., Sakthisekaran, D., Sethi, G., & Nishigaki, I. (2013). The vascular endothelium and human diseases. International journal of biological sciences, 9(10), 1057–1069. https://doi.org/10.7150/ijbs.7502
- Yeo, T. W., Lampah, D. A., Tjitra, E., Gitawati, R., Kenangalem, E., Piera, K., Granger, D. L., Lopansri, B. K., Weinberg, J. B., Price, R. N., Duffull, S. B., Celermajer, D. S., & Anstey, N. M. (2009). Relationship of cell-free hemoglobin to impaired endothelial nitric oxide bioavailability and perfusion in severe falciparum malaria. The Journal of infectious diseases, 200(10), 1522–1529. https://doi.org/10.1086/644641
- Manwani, D., & Frenette, P. S. (2013). Vaso-occlusion in sickle cell disease: pathophysiology and novel targeted therapies. Blood, 122(24), 3892–3898. https://doi.org/10.1182/blood-2013-05-498311
- Carden, M. A., Fasano, R. M., & Meier, E. R. (2020). Not all red cells sickle the same: Contributions of the reticulocyte to disease pathology in sickle cell anemia. Blood reviews, 40, 100637. https://doi.org/10.1016/j.blre.2019.100637
- Venkatesan, V., Srinivasan, S., Babu, P., & Thangavel, S. (2020). Manipulation of Developmental Gamma-Globin Gene Expression: an Approach for Healing Hemoglobinopathies. Molecular and cellular biology, 41(1), e00253-20. https://doi.org/10.1128/MCB.00253-20
- Akinsheye, I., Alsultan, A., Solovieff, N., Ngo, D., Baldwin, C. T., Sebastiani, P., Chui, D. H., & Steinberg, M. H. (2011). Fetal hemoglobin in sickle cell anemia. Blood, 118(1), 19–27. https://doi.org/10.1182/blood-2011-03-325258
- Sedrak A, Kondamudi NP. Sickle Cell Disease. [Updated 2023 Aug 28]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482384/
- Inusa, B. P. D., Hsu, L. L., Kohli, N., Patel, A., Ominu-Evbota, K., Anie, K. A., & Atoyebi, W. (2019). Sickle Cell Disease-Genetics, Pathophysiology, Clinical Presentation and Treatment. International journal of neonatal screening, 5(2), 20. https://doi.org/10.3390/ijns5020020
- Ashorobi D, Ramsey A, Yarrarapu SNS, et al. Sickle Cell Trait. [Updated 2022 Jul 18]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan- Available from: https://www.ncbi.nlm.nih.gov/books/NBK537130/
- Tran, H., Gupta, M., & Gupta, K. (2017). Targeting novel mechanisms of pain in sickle cell disease. Hematology. American Society of Hematology. Education Program, 2017(1), 546–555. https://doi.org/10.1182/asheducation-2017.1.546
- Collins, P. J., Renedo, A., & Marston, C. A. (2022). Communicating and understanding pain: Limitations of pain scales for patients with sickle cell disorder and other painful conditions. Journal of health psychology, 27(1), 103–118. https://doi.org/10.1177/1359105320944987
- Mangla A, Ehsan M, Agarwal N, et al. Sickle Cell Anemia. [Updated 2023 Sep 4]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482164/
- Kato, G. J., McGowan, V., Machado, R. F., Little, J. A., Taylor, J., 6th, Morris, C. R., Nichols, J. S., Wang, X., Poljakovic, M., Morris, S. M., Jr, & Gladwin, M. T. (2006). Lactate dehydrogenase as a biomarker of hemolysis-associated nitric oxide resistance, priapism, leg ulceration, pulmonary hypertension, and death in patients with sickle cell disease. Blood, 107(6), 2279–2285. https://doi.org/10.1182/blood-2005-06-2373
- Hankins, J. S., Penkert, R. R., Lavoie, P., Tang, L., Sun, Y., & Hurwitz, J. L. (2016). Original Research: Parvovirus B19 infection in children with sickle cell disease in the hydroxyurea era. Experimental biology and medicine (Maywood, N.J.), 241(7), 749–754. https://doi.org/10.1177/1535370216636723
- Sahu T, Pande B, Verma HK, Bhaskar LVKS, Sinha M, Sinha R, Rao PV. Infection and Potential Challenge of Childhood Mortality in Sickle Cell Disease: A Comprehensive Review of the Literature from a Global Perspective. Thalassemia Reports. 2023; 13(3):206-229. https://doi.org/10.3390/thalassrep13030019
- Friend A, Settelmeyer TP, Girzadas D. Acute Chest Syndrome. [Updated 2023 Nov 25]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan- Available from: https://www.ncbi.nlm.nih.gov/books/NBK441872/
- Ochocinski, D., Dalal, M., Black, L. V., Carr, S., Lew, J., Sullivan, K., & Kissoon, N. (2020). Life-Threatening Infectious Complications in Sickle Cell Disease: A Concise Narrative Review. Frontiers in pediatrics, 8, 38. https://doi.org/10.3389/fped.2020.00038
- Kane, I., Kumar, A., Atalla, E., & Nagalli, S. (2023). Splenic Sequestration Crisis. In StatPearls. StatPearls Publishing.
- Araujo A.N. Acute splenic sequestration in children with sickle cell anemia. J. Pediatr. (Rio J.) 2009; 85:373–374. doi: 10.1590/S0021-75572009000400018.
- Parikh, T., Goti, A., Yashi, K., Gopalakrishnan Ravikumar, N. P., Parmar, N., Dankhara, N., & Satodiya, V. (2023). Pediatric Sickle Cell Disease and Stroke: A Literature Review. Cureus, 15(1), e34003. https://doi.org/10.7759/cureus.34003
- Dowling, M. M., Quinn, C. T., Rogers, Z. R., & Buchanan, G. R. (2010). Acute silent cerebral infarction in children with sickle cell anemia. Pediatric blood & cancer, 54(3), 461–464. https://doi.org/10.1002/pbc.22242
- Pan, Y., Wan, W., Xiang, M., & Guan, Y. (2022). Transcranial Doppler Ultrasonography as a Diagnostic Tool for Cerebrovascular Disorders. Frontiers in human neuroscience, 16, 841809. https://doi.org/10.3389/fnhum.2022.841809
- Shah, R., Taborda, C., & Chawla, S. (2017). Acute and chronic hepatobiliary manifestations of sickle cell disease: A review. World journal of gastrointestinal pathophysiology, 8(3), 108–116. https://doi.org/10.4291/wjgp.v8.i3.108
- Crane, G. M., & Bennett, N. E., Jr (2011). Priapism in sickle cell anemia: emerging mechanistic understanding and better preventative strategies. Anemia, 2011, 297364. https://doi.org/10.1155/2011/297364
- Gragg J, Blair K, Baker MB. Hyphema. [Updated 2022 Dec 26]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK507802/
- Farris W, Waymack JR. Central Retinal Artery Occlusion. [Updated 2023 Sep 4]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK470354/
- Dagra, A., Lucke–Wold, B., McGrath, K., Mehkri, I., Mehkri, Y., Davidson, C. G., Gilberstadt, N., Douglas, B. H., & Hoh, B. L. (2024). Central Retinal Artery Occlusion: A review of pathophysiological features and management. Stroke: Vascular and Interventional Neurology, 4(1). https://doi.org/10.1161/svin.123.000977
- Mohankumar A, Gurnani B. Orbital Apex Syndrome. [Updated 2023 Jun 11]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK592386/
- Al-Suleiman A. M. (2006). Diagnosis: Orbital Compression Syndrome in Sickle Cell Disease. Annals of Saudi Medicine, 26(1), 76. https://doi.org/10.5144/0256-4947.2006.76
- Bonanomi, M. T. B. C., & Lavezzo, M. M. (2013). Sickle cell retinopathy: diagnosis and treatment. Arquivos Brasileiros De Oftalmologia, 76(5), 320–327. https://doi.org/10.1590/s0004-27492013000500016
- Soliman, A. T., De Sanctis, V., Yassin, M. A., Alshurafa, A., Ata, F., & Nashwan, A. J. (2022). Blood transfusion and iron overload in patients with Sickle Cell Disease (SCD): Personal experience and a short update of diabetes mellitus occurrence. PubMed, 93(4), e2022291. https://doi.org/10.23750/abm.v93i4.13330
- Rasel M, Mahboobi SK. Transfusion Iron Overload. [Updated 2023 Apr 2]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK562146/
- Brittenham GM, Griffith PM, Nienhuis AW, McLaren CE, Young NS, Tucker EE, Allen CJ, Farrell DE, Harris JW. Efficacy of deferoxamine in preventing complications of iron overload in patients with thalassemia major. N Engl J Med. 1994 Sep 01;331(9):567-73
- Bedair, E., Almaslamani, N., & Yassin, M. A. (2023). Radiological manifestation of avascular necrosis (AVN) in sickle cell disease (SCD): a review of diagnostic imaging. PubMed, 94(3), e2023177. https://doi.org/10.23750/abm.v94i3.14714
- Belisário, A. R., Mendes-Oliveira, F., De Souza, V. R., Bolina-Santos, E., Mendes, F. G., Moreno, E. C., Franca, A. T., Sabino, É. C., Otta, D. A., De Faria, E. S., Coelho-Dos-Reis, J. G. A., Martins‐Filho, O. A., & De Freitas Carneiro-Proietti, A. B. (2022). Association between inflammatory molecules, nitric oxide metabolites and leg ulcers in individuals with sickle cell anemia. Hematology, Transfusion and Cell Therapy, 44(2), 169–176. https://doi.org/10.1016/j.htct.2020.09.152
- Santos, E. D. C., Santana, P. V. B., De Jesus, L. L. S., Melo, G. I. V., Yahouédéhou, S. C. M. A., Da Guarda, C. C., Santiago, R. P., Fiuza, L. M., Carvalho, S. P., Santos, L. O. D., Adôrno, E. V., Aleluia, A. C. M., Campos, L. C. G., Fonseca, T. C., De Souza Gonçalves, M., & Aleluia, M. M. (2023). Leg ulcers in sickle cell disease: A multifactorial analysis highlights the hemolytic profile. Hematology Reviews, 15(1), 119–129. https://doi.org/10.3390/hematolrep15010013
- Hayes, M. M., Vedamurthy, A., George, G., Dweik, R. A., Klings, E. S., Machado, R. F., Gladwin, M. T., Wilson, K. C., & Thomson, C. C. (2014). Pulmonary hypertension in sickle cell disease. Annals of the American Thoracic Society, 11(9), 1488–1489. https://doi.org/10.1513/annalsats.201408-405cme
- Nath, K. A., & Hebbel, R. P. (2015). Sickle cell disease: renal manifestations and mechanisms. Nature Reviews Nephrology, 11(3), 161–171. https://doi.org/10.1038/nrneph.2015.8
- Aeddula, N. R. (2023, September 4). Sickle cell nephropathy. StatPearls – NCBI Bookshelf. https://www.ncbi.nlm.nih.gov/books/NBK526017/
- Obadina, M., Wilson, S., Derebail, V. K., & Little, J. A. (2023). Emerging Therapies and Advances in Sickle Cell Disease with a Focus on Renal Manifestations. Kidney360, 4(7), 997–1005. https://doi.org/10.34067/kid.0000000000000162
- Mulumba, L. L., & Wilson, L. (2015). Sickle cell disease among children in Africa: An integrative literature review and global recommendations. International Journal of Africa Nursing Sciences, 3, 56–64. https://doi.org/10.1016/j.ijans.2015.08.002
- Tezol, O., Karahan, F., & Unal, S. (2021). Sickle Cell Disease and Psychosocial Well-Being: Comparison of Patients With Preclinical and Clinical Avascular Necrosis of the Femoral Head. Turkish archives of pediatrics, 56(4), 308–315. https://doi.org/10.5152/TurkArchPediatr.2021.20270
- Anie K.A., Green J. Psychological therapies for sickle cell disease and pain. Cochrane Database Syst. Rev. 2015 doi: 10.1002/14651858.CD001916
- Sickle Cell Society Standards for the Clinical Care of Adults with Sickle Cell Disease in the UK—2018
- Mayo Clinic. (2024). Sickle cell anemia: Diagnosis & treatment. Retrieved from https://www.mayoclinic.org/diseases-conditions/sickle-cell-anemia/diagnosis-treatment/drc-20355882
- Office of the Commissioner. (2023, December 8). FDA Approves First Gene Therapies to Treat Patients with Sickle Cell Disease. U.S. Food And Drug Administration. https://www.fda.gov/news-events/press-announcements/fda-approves-first-gene-therapies-treat-patients-sickle-cell-disease
- Childerhose, J. E., Cronin, R. M., Klatt, M. D., & Schamess, A. (2023). Treating chronic pain in sickle cell disease — the need for a biopsychosocial model. The New England Journal of Medicine, 388(15), 1349–1351. https://doi.org/10.1056/nejmp2301143
- Telfer, P., & Kaya, B. (2017). Optimizing the care model for an uncomplicated acute pain episode in sickle cell disease. Hematology, 2017(1), 525–533. https://doi.org/10.1182/asheducation-2017.1.525
- Yale, S. H., Nagib, N., & Guthrie, T. (2000, March 1). Approach to the Vaso-occlusive Crisis in Adults with Sickle Cell Disease. AAFP. https://www.aafp.org/pubs/afp/issues/2000/0301/p1349.html
- Rees, D. C., Brousse, V., & Brewin, J. (2022). Determinants of severity in sickle cell disease. Blood Reviews, 56, 100983. https://doi.org/10.1016/j.blre.2022.100983
- Bedair, E., Almaslamani, N., & Yassin, M. A. (2023). Radiological manifestation of avascular necrosis (AVN) in sickle cell disease (SCD): a review of diagnostic imaging. PubMed, 94(3), e2023177. https://doi.org/10.23750/abm.v94i3.14714
- Detterich, J. (2018). Simple chronic transfusion therapy, a crucial therapeutic option for sickle cell disease, improves but does not normalize blood rheology: What should be our goals for transfusion therapy? Clinical Hemorheology and Microcirculation, 68(2–3), 173–186. https://doi.org/10.3233/ch-189006
- Meliti, A., Muftah, S., Saleh, D., & Habibullah, N. (2023). Compound Heterogeneous Sickle Cell-B+ Thalassemia Incidentally Discovered Through Cytological Examination of a Fine-Needle Aspiration Specimen from an Aneurysmal Bone Cyst in a Young Child: A Case Report. Cureus. https://doi.org/10.7759/cureus.33594
- Purnell, M. C., & Rayborn, M. (2020). Novel hydration and nutritional strategies for sickle cell disease. EJHaem, 1(1), 230–234. https://doi.org/10.1002/jha2.9
- Rolle, A., Vidal, E., Laguette, P., Garnier, Y., Delta, D., Martino, F., Portecop, P., Etienne‐Julan, M., Piednoir, P., De Jong, A., Romana, M., & Bernit, E. (2023). Pain control for sickle cell crisis, a novel approach? A Retrospective study. Medicina-lithuania, 59(12), 2196. https://doi.org/10.3390/medicina59122196
- Zouki, T., Haroutunian, A., & Malcolm, T. L. (2018). Pain management for the sickle cell patient. In IntechOpen eBooks. https://doi.org/10.5772/intechopen.79495
- Arzoun, H., Srinivasan, M., Sahib, I., Fondeur, J., Mendez, L. E., Hamouda, R. K., & Mohammed, L. (2022). Opioid use in patients with sickle cell disease during a Vaso-Occlusive Crisis: a systematic review. Cureus. https://doi.org/10.7759/cureus.21473
- Ortiz, F. O., Aldrich, T. K., Nagel, R. L., & Benjamin, L. J. (1999). Accuracy of pulse oximetry in sickle cell disease. American Journal of Respiratory and Critical Care Medicine, 159(2), 447–451. https://doi.org/10.1164/ajrccm.159.2.9806108
- Indications for RBC Exchange Transfusion in Patients with Sickle Cell Disease: Revisited. (2019, November 1). PubMed. https://pubmed.ncbi.nlm.nih.gov/31882437/
- Howard, J. (2016). Sickle cell disease: when and how to transfuse. Hematology, 2016(1), 625–631. https://doi.org/10.1182/asheducation-2016.1.625
- Okomo, U., & Meremikwu, M. (2017). Fluid replacement therapy for acute episodes of pain in people with sickle cell disease. The Cochrane Library, 2021(4). https://doi.org/10.1002/14651858.cd005406.pub5
- Rai, P., & Ataga, K. I. (2020). Drug therapies for the management of sickle cell disease. F1000Research, 9, 592. https://doi.org/10.12688/f1000research.22433.1
- Da Guarda, C. C., Silveira-Mattos, P. S., Yahouédéhou, S. C. M. A., Santiago, R. P., Aleluia, M. M., Figueiredo, C. V. B., Fiuza, L. M., Carvalho, S. P., De Oliveira, R. M., Nascimento, V. M. L., Luz, N. F., Borges, V. M., Andrade, B. B., & Gonçalves, M. S. (2019). Hydroxyurea alters circulating monocyte subsets and dampens its inflammatory potential in sickle cell anemia patients. Scientific Reports, 9(1). https://doi.org/10.1038/s41598-019-51339-x
- Wu, H., Gannon, M., & Hsu, L. L. (2022). Evaluation of glutamine utilization in patients with sickle cell disease. Journal of Pediatric Hematology Oncology, 45(1), e52–e55. https://doi.org/10.1097/mph.0000000000002519
- Oksenberg, D., Dufu, K., Patel, M., Chuang, C., Li, Z., Xu, Q., Silva-García, A., Zhou, C., Hutchaleelaha, A., Patskovska, L., Patskovsky, Y., Almo, S. C., Sinha, U. S. P., Metcalf, B. W., & Archer, D. (2016). GBT440 increases haemoglobin oxygen affinity, reduces sickling, and prolongs RBC half‐life in a murine model of sickle cell disease. British Journal of Haematology, 175(1), 141–153. https://doi.org/10.1111/bjh.14214
- Daak, A., López‐Toledano, M. A., & Heeney, M. M. (2020). Biochemical and therapeutic effects of Omega-3 fatty acids in sickle cell disease. Complementary Therapies in Medicine, 52, 102482. https://doi.org/10.1016/j.ctim.2020.102482
- Dixit, R., Nettem, S., Singh, S., Soe, H. H. K., Abas, A. B., Vance, L. D., & Stover, P. J. (2018). Folate supplementation in people with sickle cell disease. The Cochrane Library, 2021(4). https://doi.org/10.1002/14651858.cd011130.pub3
- Esoh, K., & Wonkam, A. (2021). Evolutionary history of sickle-cell mutation: implications for global genetic medicine. Human Molecular Genetics, 30(R1), R119–R128. https://doi.org/10.1093/hmg/ddab004
- National Academies Press (US). (2020, September 10). Societal and structural contributors to disease impact. Addressing Sickle Cell Disease – NCBI Bookshelf. https://www.ncbi.nlm.nih.gov/books/NBK566460/
- Padda, A., Corriveau‐Bourque, C., Belletrutti, M., & Bruce, A. (2019). Supplemental oxygen therapy recommendations in patients with sickle cell disease during air travel: A cross-sectional survey of North American health care providers. Paediatrics and Child Health, 25(2), 107–112. https://doi.org/10.1093/pch/pxz049
- Jonk, A. M., Van Den Berg, I. P., Olfert, I. M., Wray, D. W., Arai, T., Hopkins, S. R., & Wagner, P. D. (2007). Effect of acetazolamide on pulmonary and muscle gas exchange during normoxic and hypoxic exercise. The Journal of Physiology, 579(3), 909–921. https://doi.org/10.1113/jphysiol.2006.120949
Disclaimer:
Use of Course Content. The courses provided by NCC are based on industry knowledge and input from professional nurses, experts, practitioners, and other individuals and institutions. The information presented in this course is intended solely for the use of healthcare professionals taking this course, for credit, from NCC. The information is designed to assist healthcare professionals, including nurses, in addressing issues associated with healthcare. The information provided in this course is general in nature and is not designed to address any specific situation. This publication in no way absolves facilities of their responsibility for the appropriate orientation of healthcare professionals. Hospitals or other organizations using this publication as a part of their own orientation processes should review the contents of this publication to ensure accuracy and compliance before using this publication. Knowledge, procedures or insight gained from the Student in the course of taking classes provided by NCC may be used at the Student’s discretion during their course of work or otherwise in a professional capacity. The Student understands and agrees that NCC shall not be held liable for any acts, errors, advice or omissions provided by the Student based on knowledge or advice acquired by NCC. The Student is solely responsible for his/her own actions, even if information and/or education was acquired from a NCC course pertaining to that action or actions. By clicking “complete” you are agreeing to these terms of use.
➁ Complete Survey
Give us your thoughts and feedback
➂ Click Complete
To receive your certificate